Что нельзя при нейрофиброматозе
Согласно классификации В. Риккарди (1982), выделяют семь типов заболевания.
Наиболее часто (85–97% всех случаев) встречается нейрофиброматоз типа I. В настоящее время доказана генетическая самостоятельность нейрофиброматоза типа I, II:
Нейрофиброматоз I типа встречается среди представителей всех рас, с частотой 1 на 3 000 новорожденных; 20–40 на 100 000 населения в общей популяции. Оба пола поражаются одинаково часто.
Согласно теории нейрокристопатии, патогенез заболевания связан с поражением эмбрионального нервного валика, приводящим к нарушению миграции происходящих из него клеток, в частности шванновских клеток и меланоцитов, их роста и дифференцировки на различных стадиях развития организма.
Пятна «кофе с молоком»
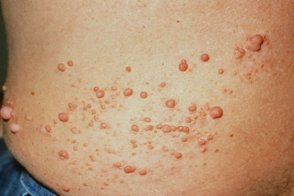
Нейрофибромы
Плексиформные нейрофибромы
Другие кожные симптомы
Глазные симптомы
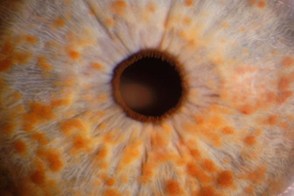 Из внекожных симптомов наибольшее диагностическое значение имеют пигментные гамартомы радужной оболочки (узелки Лиша), возникающие в детском возрасте и обнаруживаемые с помощью щелевой лампы. Частота их выявления нарастает с возрастом. Из других поражений глаз часто встречаются глиомы зрительного нерва. Из внекожных симптомов наибольшее диагностическое значение имеют пигментные гамартомы радужной оболочки (узелки Лиша), возникающие в детском возрасте и обнаруживаемые с помощью щелевой лампы. Частота их выявления нарастает с возрастом. Из других поражений глаз часто встречаются глиомы зрительного нерва. |
Патология нервной системы
Патология костной системы
При тяжелом течении болезни дисплазия позвонков сочетается с параспинальными нейрофибромами, при росте которых разрушаются прилежащие участки позвоночного столба. У большинства больных развивается сколиоз, обычно на втором десятилетии жизни. Обычно поражается шейно-грудной отдел позвоночника. Сколиоз, как правило, прогрессирует, и в сочетании с выраженным кифозом нередко приводит к неврологическим и сердечно-легочным осложнениям. Характерны псевдоартрозы трубчатых костей, особенно большеберцовых. Обычно эта патология врожденная.
Эндокринные нарушения
Эндокринные нарушения проявляются акромегалией, сахарным диабетом, гипогонадизмом или, наоборот, преждевременным половым созреванием. Наблюдают недостаточность надпочечников, щитовидной железы, кретинизм, гиперпаратироидизм, но самой частой патологией считают феохромоцитому.
Патология внутренних органов
Диагностическим критерием заболевания типа I считают наличие двух или более признаков из указанных ниже (ВОЗ, 1992 г.):
При нейрофиброматозе типа I возможна пренатальная диагностика, а также предимплантационная диагностика в процессе проведения ЭКО для семейных пар с высоким риском передачи патологического гена.
Диагноз «нейрофиброматоз центрального типа» может быть поставлен при наличии одного из следующих критериев (ВОЗ, 1992):
При гистологическом исследовании шванномы представлены инкапсулированными опухолями, состоящими из вытянутых веретенообразных клеток и фибриллярного эозинофильного межклеточного матрикса. Участки скопления параллельных рядов клеток в виде частокола, отделенные друг от друга бесклеточным пространством, носят название зоны Антони А, при этом формируются характерные тельца Верокая. Участки отечной муцинозной стромы названы зоной Антони В.
Дифференциальную диагностику проводят с другими формами нейрофиброматоза. Возможна пренатальная диагностика нейрофиброматоза типа II, а также предимплантационная диагностика в процессе проведения ЭКО для семейных пар с высоким риском передачи патологического гена.
Нейрофиброматоз типа IV (вариантный) также сходен с центральным нейрофиброматозом, однако отличается более многочисленными кожными нейрофибромами, высоким риском развития глиомы зрительного нерва, нейролеммом и менингиом.
Особо выделяют сегментарный нейрофиброматоз (тип V). Для него характерно одностороннее поражение (нейрофибромы и/или пигментные пятна) какого-либо сегмента тела или его части. Полагают, что развитие этой формы нейрофиброматоза связано с соматической мутацией. Клиническая картина может напоминать таковую при гемигипертрофии; возможно развитие изолированного псевдоартроза конечности; изолированной глиомы зрительного нерва; изолированной плексиформной нейрофибромы. При этом узелки Лиша обнаруживают ипсилатерально. Если они двусторонние, полагают, что данное состояние не отвечает критериям сегментарного нейрофиброматоза. Описан двусторонний сегментарный нейрофиброматоз.
При нейрофиброматозе типа VI нейрофибромы отсутствуют, имеются только пигментные пятна. Диагностика этого заболевания сложна, если не появляются новые случаи в последующих поколениях.
Поздний нейрофиброматоз (тип VII) возникает после 20 лет. По мнению некоторых авторов если процесс развивается в третьей декаде жизни на одном участке тела или более, то он не ассоциирован с пигментными пятнами, узелками Лиша, псевдоартрозом и нарушением умственного развития. Семейные случаи неизвестны.
Отдельные, наиболее крупные нейрофибромы могут быть удалены хирургически. Это показано главным образом при подозрении на злокачественный процесс. Однако после удаления нейрофибром, особенно плексиформных, нередки рецидивы.
Методика лечения включает препараты, влияющие на:
Продолжительность каждого курса лечения составляет около 3 мес.
Показано медико-генетическое консультирование. Риск унаследовать заболевание для детей больных составляет 50%, независимо от степени клинических проявлений у пробанда. При классическом варианте около 10% носителей гена могут не иметь клинических проявлений. При II типе заболевания пенетрантность более высокая.
Прогноз для выздоровления неблагоприятный, его тяжесть определяется степенью вовлечения в процесс внутренних органов и систем.
Часто задаваемые вопросы
Можно ли вылечить нейрофиброматоз?
Поскольку нейрофиброматоз является генетическим заболеванием, на сегодняшний день, лечение исключительно симптоматическое, однако проводятся исследования в области генной терапии, а также в апреле 2020 года в США было одобрено первое в мире лекарство для лечения неоперабельных плексиформных нейрофиброма для пациентов с нейрофиброматозом 1 типа.
Может ли один тип нейрофиброматоза перетекать в другой?
Нет, это невозможно. Все три типа заболевания (нейрофиброматоз 1 типа, нейрофиброматоз 2 типа и шванноматоз) по сути являются разными заболеваниями.
Существует ли возможность предсказать течение заболевания?
Симптомы и тяжесть течения нейрофиброматоза 1 типа варьируются от человека к человеку и, к сожалению, не могут быть предсказаны заранее врачами в полной мере даже при наследственном типе заболевания.
При наследовании нейрофиброматоза 2 типа, симптомы ребенка часто похожи на симптомы родителя с НФ2. При новой мутации НФ2 у человека возможно в определенной степени предсказать течение заболевания основываясь на поврежденные гены.
Течение шванноматоза на сегодняшний день предсказать невозможно.
Можно ли пациентам с нейрофиброматозом делать массаж, ЛФК, физиотерапию?
Диагноз нейрофиброматоз сам по себе не является противопоказанием к массажу, физиотерапии, ЛФК, нахождению на солнце и тд. Нет подтвержденных данных о вреде указанных процедур для пациентов с нейрофиброматозом. Однако, в ряде случаев, лечащий врач может запретить процедуры (например, при ложном суставе, прохождении химиотерапии и т.д.).
Можно ли детям с нейрофиброматозом ставить прививки?
Нейрофиброматоз не является противопоказанием к прививанию, однако, лечащий врач, в некоторых случаях может отложить или запретить постановку прививки (например, в случае обнаружения опухолей, прохождении химиотерапии и т.д.)
Нейрофиброматоз
Содержание статьи:
Каждому человеку хотя бы раз в жизни приходилось встречать людей, кожа которых покрыта пигментными пятнами и опухолеподобными разрастаниями. Скорее всего, вы увидели больного, страдающего нейрофибоматозом. Это одно из часто встречающихся наследственных заболеваний. Оно требует постоянного наблюдения и лечения у невролога, а также других специалистов. Иногда больному требуются проведение хирургических вмешательств.
Описание
Нейрофиброматоз – это заболевание относят к группе наследственных системных заболеваний. Оно принадлежит к группе факоматозов. При этих патологиях происходит сочетанное поражение кожи и структур нервной системы. Для него типичны нарушение развития эктодермальных и мезодермальных структур. К ним относят: кожу, нервную систему и скелет. Клинические проявления болезни могут появиться в любом возрасте.
Выделяют шесть типов нейрофиброматоза. Из них чаще встречаются и имеют большую клиническую значимость нейрофиброматоз I типа (болезнь Реклингхаузена), а также нейрофиброматоз II типа (нейрофиброматоз с невриномами VIII пары черепных нервов). Остальные формы встречаются довольно редко.
При этом заболевании образуются множественные нейрофибромы. Это опухоли доброкачественного характера. Они развиваются их клеток, составляющих оболочки нервных волокон. Эти образования чаще всего расположены в толще кожи и подкожной клетчатке, а также в межмышечном (мейсснеровом) и подслизистом (ауэрбаховом) пространстве.
Изредка они находятся в головном мозге, поражают нервные волокна, спинномозговые корешки, внутренние органы и мягкие ткани. На коже у больных появляются множественные пигментные пятна. У этих больных повышен риск развития злокачественных образований.
Причины
Нейрофиброматоз первого типа имеет наследственный характер, передается по аутосомно-доминантному типу наследования с высокой степенью пенетрантности. Оно одинаково часто встречается как у мужчин, так и у женщин. Ген НФ1, кодирующий заболевание, расположен в 17-й хромосоме на ее длинном плече. Он подавляет рост опухолевых тканей. При его дефекте отмечают нарушение синтеза белков, которые ответственны за размножение клеток.
Случаи заболевания нейрофибоматозом первого типа встречается примерно у одного ребенка на 3500 новорождённых. Эта патология обычно сочетается с развитием аномалий других органов и систем. Риск наследования при наличии болезни у одного из родителей составляет 50%. Если оно выявлено у обоих родителей, возможность рождения больного малыша составляет примерно 75%.
Причиной нейрофиброматоза второго типа считают мутацию гена НФ2, находящегося на 22 хромосоме. Он ответственен за продукцию белка мерлина, который является супрессором роста опухолей. Для этого заболевания характерен аутосомно-доминантный тип наследования с небольшой степенью пенетрантности. Другие типы нейрофиброматоза тоже связаны с мутациями в генах, кодирующих различные белки, регулирующие опухолевый рост.
Симптомы
Выраженность клинических проявлений при нейрофиброматозе обусловлен степенью поражения гена и недостаточностью выработки беков, подавляющих опухолевый рост. Самым характерным симптомом для нейрофиброматоза является гиперпигментация. У детей при рождении или несколько позже формируются пятна. Их цвет бывает разной интенсивности от светлого до темно-коричневого цвета, изредка они окрашены в багровый цвет.
Узелки Лиша (гамартомы) считают специфическим признаком нейрофиброматоза I типа. Их находят на радужке глаза. Они представляют собой пигментные пятна небольшого размера. Эти образования белесоватого или светло-бежевого оттенка. Они заметны только офтальмологу при специальном осмотре. Их становится больше по мере взросления человека.
В пубертатный период начинают появляться плексиформные и кожные нейрофибромы. Они располагаются под кожей, по ходу крупных нервов. Это мелкие доброкачественные опухоли, представляющие собой не более чем косметический дефект. Плексиформные образования, расположенные по длине периферических нервов, появляются на веках, конъюнктиве, в средостении, брюшной полости. Проявляются постоянной болью, онемением кожи, судорогами, параличами.
При этом заболевании опухоли ЦНС локализуются в полости черепа. Они представлены различными новообразованиями: глиомы зрительных нервов, астроцитомы, эпендимомы, невриномы слуховых нервов, менингиомы, нейрофибромы. Клинику определяет размер новообразований, вовлеченностью структур нервной системы в процесс. В детстве у этих больных выявляют расстройства развития: снижение интеллекта, гиперактивность.
При тяжелом течении болезни происходит деформация костей. У больного формируется сколиоз. Развиваются атрофии или гипертрофии трубчатых костей. Они искривляются, на поверхности их разрастаются гребни и наслоения. В костных полостях образуются нейрофибромы. При вовлечении в процесс костей черепа выявляют внешнюю асимметрию, которая выражена при поражении лица и глазниц. На своде черепа возникают участки атрофий, а изредка отмечают костные разрастания.
При нейрофиброматозе II типа образуются высокодифференцированные опухоли. Они ведут себя агрессивнее, чем при I типе заболевания.
Заметили у себя один или несколько симптомов?
Записаться на прием
Диагностика
Обследование больного для установления диагноза ведется группой специалистов. В нее входят: невролог, дерматовенеролог, офтальмолог, генетик, отоларинголог. У больного выясняют жалобы, его опрашивают с целью выяснения семейного анамнеза заболевания. При осмотре выявляют характерные поражения кожи, костей, нервной системы, внутренних органов.
Для уточнения диагноза проводят:
Участки опухолей берут на биопсию. Затем проводится гистологическое исследование биоптата.
Лечение
Часто в ведении больных с нейрофиброматозом ограничиваются симптоматическим лечением. Если нейрофибромы расположены в местах повышенного травмирования и провоцируют боль, их удаляют хирургическими методами. При множественных образованиях назначают химиотерапию, проводят облучение. При поражении опорно-двигательного аппарата проводят реабилитационные мероприятия.
Заболевание имеет благоприятный прогноз. Опухолевые образования озлокачествляется крайне редко. При выполнении специальных мероприятий у больных сохраняется удовлетворительное качество жизни и работоспособность.
Врачи отделения
Табельский Роман Александрович
Врач невролог, врач нейрохирург, высшая врачебная квалификационная категория, стаж работы 17 лет
Архив
НЕЙРОФИБРОМАТОЗ
Нейрофиброматоз (НФ) впервые был признан отдельной нозологией в конце 19-го века, когда Фридрих фон Реклингаузен описал серию пациентов с сочетанием кожных поражений и опухолями центральной и периферической нервной системы. Окончательная дифференциальная диагностика между 1 и 2 типом заболевания (НФ1 и НФ2) была проведена в 1981 году. Данные заболевания значительно отличаются, тем не менее, некоторые их симптомы подобны, а это часто приводит к сложностям в дифференциальной диагностике и оценке динамики состояния. Представленный обзор посвящен характерным симптомам двух упомянутых клинических единиц, их патогенезу, диагностике и терапии.
ГЕНЕТИКА И ПАТОГЕНЕЗ
НФ1 и НФ2 являются аутосомно-доминантными генетическими заболеваниями без какого-либо расового или полового преобладания. Их локусы расположены, соответственно, на хромосомах 17q11.2 и 22q12.2. Для указанных заболеваний типична высокая частота спонтанных мутаций, в результате чего 50% клинических случаев являются спорадическими. Оба заболевания характеризуются 100% пенетрантностью и широкой фенотипической вариабельностью. Факторы последней до сих пор непонятны, они, вероятно, могут быть как наследственными, так и экзогенными. НФ1 довольно распространен, его частота составляет примерно 1:3000. Частота НФ2 равняется 1:40000. Для обоих состояний характерна генетическая мозаичность.
Следствием мутаций гена НФ1 является утрата функции белка нейрофибромина, способствующего удержанию специфического протоонкогена в неактивной форме. В результате этого в тканях ЦНС и кожи начинается неуправляемая пролиферация, лежащая в основе туморогенеза. Ген НФ2 кодирует белок мерлин, регулирующий рост, мобильность и клеточное ремоделирование путем угнетения передачи внеклеточных митогенных сигналов.
Генетическое тестирование доступно для обеих форм НФ. Его чувствительность колеблется в диапазоне 34–95%. Соматическая мозаичность, как правило, связана с гиподиагностикой ненаследственных форм. В таких случаях оправданным является анализ характерных для семьи мутаций, что ассоциируется с достаточно высокой диагностической точностью.
КЛИНИЧЕСКИЕ ПРИЗНАКИ: ОБЗОР
НФ1 характеризуется преимущественно кожными проявлениями (гиперпигментированными макулами цвета кофе с молоком, кожными и подкожными нейрофибромами), опухолями невральных оболочек, глиомами зрительного тракта (ГЗТ) и другими нейроонкологическими заболеваниями, целым рядом костных аномалий, когнитивным дефицитом и повышенным риском опухолевого роста за пределами нервной ткани. Для НФ2 характерны новообразования центральной и периферической нервной системы (в частности шванномы) при минимальных кожных и экстраневральных симптомах (табл. 1). Оба заболевания диагностируются на основании клинических признаков (табл. 2, 3, 4).
| НФ1 | НФ2 | |
|---|---|---|
| Тип наследования | Аутосомно-доминантный | Аутосомно-доминантный |
| Спонтанные мутации | 50% | 50% |
| Частота | 1 : 3000 | 1 : 40000 |
| Вовлеченные хромосомы | 17q11.2 | 22q12.2 |
| Генный продукт | Нейрофибромин | Мерлин |
| Типичные признаки | Макулы цвета кофе с молоком в детстве (включая раннее) | Глухота, вестибулярные нарушения (у молодых лиц) и катаракта |
| Опухоли нервных оболочек | Нейрофиброма, плексиформная нейрофиброма, злокачественные опухоли периферических нервов | Шваннома |
| Внутричерепные опухоли | Глиомы зрительных путей, другие астроцитомы/глиомы | Вестибулярные шванномы, менингиомы |
| Спинномозговые опухоли | Узловые плексиформные нейрофибромы (корешков) | Шванномы (корешков), эпендимомы (интрамедуллярные) |
| Кожные симптомы | Макулы цвета кофе с молоком, веснушки в паховых/подмышечных областях, кожные/подкожные нейрофибромы | Кожные шванномы |
| Когнитивные расстройства | Часто встречаются трудности в учебе, синдром нарушения внимания и гиперактивности, легкое снижение IQ | Отсутствуют |
| Скелетные проявления | Сколиоз, сутулость, псевдоартрозы, дисплазия клиновидной кости | Отсутствуют |
| Офтальмологические нарушения | Узелки Лиша, врожденная глаукома | Ювенильное подкапсулярное помутнение хрусталика, катаракта, рубцы роговицы, гамартомы сетчатки |
| Другие типы опухолей | Хроническая миелогенная лейкемия, феохромоцитома | Отсутствуют |
| Другие неврологические проявления | Макроэнцефалия, невыясненные зоны прояснения (на МРТ) | Невропатии, арефлексия |
| Другие симптомы | Сосудистые аномалии, желудочно-кишечные кровотечения, запоры |
| НФ1 диагностируется у пациента при наличии 2 и более из нижеперечисленных признаков: |
|---|
| НФ2 диагностируется у пациента при наличии какого-либо из нижеперечисленных симптомов: |
|---|








